
Caratteristiche
La mastocitosi sistemica (SM) è una malattia rara causata dall’espansione clonale e dall’accumulo di mastociti anomali in tutto il corpo, in diversi organi e tessuti. Si tratta di una malattia complessa e invalidante che influenza negativamente la qualità della vita del Paziente.
Che cosa sono i mastociti?
Sono cellule del sistema immunitario che hanno origine nel midollo osseo a partire da una cellula staminale emopoietica (capostipite di tutti gli elementi del sangue) pluripotente.
Il mastocita svolge un ruolo cruciale nelle reazioni allergiche e nell’infiammazione.

Cosa provoca la proliferazione e l’accumulo dei mastociti?
Le mutazioni a carico del gene c-KIT innescano
l’attivazione del recettore KIT anche in assenza
del suo ligando.
Questo meccanismo:
- favorisce la proliferazione cellulare,
- riduce la morte programmata delle cellule e, di conseguenza, l’accumulo di mastociti in alcuni organi e tessuto del corpo umano.
Quali sono le forme principali di mastocitosi?
Ne esistono 2:
- la mastocitosi cutanea, si osserva soprattutto nei bambini e spesso si risolve durante l’adolescenza. Il quadro clinico è caratterizzato da lesioni cutanee che indicano infiltrazione e aggregati di mastociti anomali,
- la mastocitosi sistemica, che è cronica; si osserva principalmente negli adulti e generalmente colpisce il midollo osseo.
- Esistono almeno 4 sottotipi di mastocitosi sistemica: mastocitosi indolente (indolent systemic mastocytosis), mastocitosi del midollo osseo (bone marrow mastocytosis), mastocitosi smoldering (smoldering systemic mastocytosis), e mastocitosi avanzata (advanced systemic mastocytosis).
- A sua volta la mastocitosi sistemica in fase avanzata (o AdvSM), più impattante, può essere classificata come:
- la mastocitosi con una neoplasia ematologica associata (SM-AHN),
- la leucemia mastocitaria (MCL),
- la mastocitosi sistemica aggressiva (ASM).
Sintomi
Sono 3 i tipi di sintomi che possono manifestarsi nel Paziente con mastocitosi, anche a seconda della forma di malattia.
Sintomi da rilascio di mediatori:

- flushing,
- orticaria,
- episodi di ipotensione/anafilassi,
- cefalea,
- nausea, vomito e diarrea,
- crampi addominali,
- reazione avversa ad alimenti (arachidi, noci, nocciole),
- reazione avversa a farmaci (i più frequenti sono β-lattamici, FANS e anestetici),
- reazione avversa a puntura di imenotteri,
- osteoporosi severa.
Sintomi cutanei:

In caso di accumulo di sintomi confinato alla cute, come nel caso della mastocitosi cutanea:
- piccole placche cutanee disseminate (orticaria pigmentosa),
- lesione vescicolare singola (mastocitoma solitario),
- iperpigmentazione, noduli e arrossamento.
Sintomi sistemici:

Causati dall’infiltrazione dei mastociti negli organi interni, solamente presenti nella forma di mastocitosi sistemica aggressiva:
- coinvolgimento cutaneo,
- ingrossamento della milza,
- epatopatia e ascite,
- osteoporosi e riassorbimento osseo,
- reazioni allergiche ricorrenti,
- ipotensione arteriosa e incremento della frequenza,
- febbre e sudorazioni notturne,
- fatigue,
- sintomi neurologici e psichiatrici,
- diarrea,
- malattia ulcerativa gastrointestinale,
- malassorbimento intestinale,
- calo ponderale,
- alterazioni dei valori del sangue.
Diagnosi
La diagnosi di mastocitosi sistemica si basa sulla valutazione delle caratteristiche morfologiche, immunofenotipiche e genetiche dei mastociti, come riassunto dallo schema sottostante.
Ricostruzione grafica della diagnosi di mastocitosi
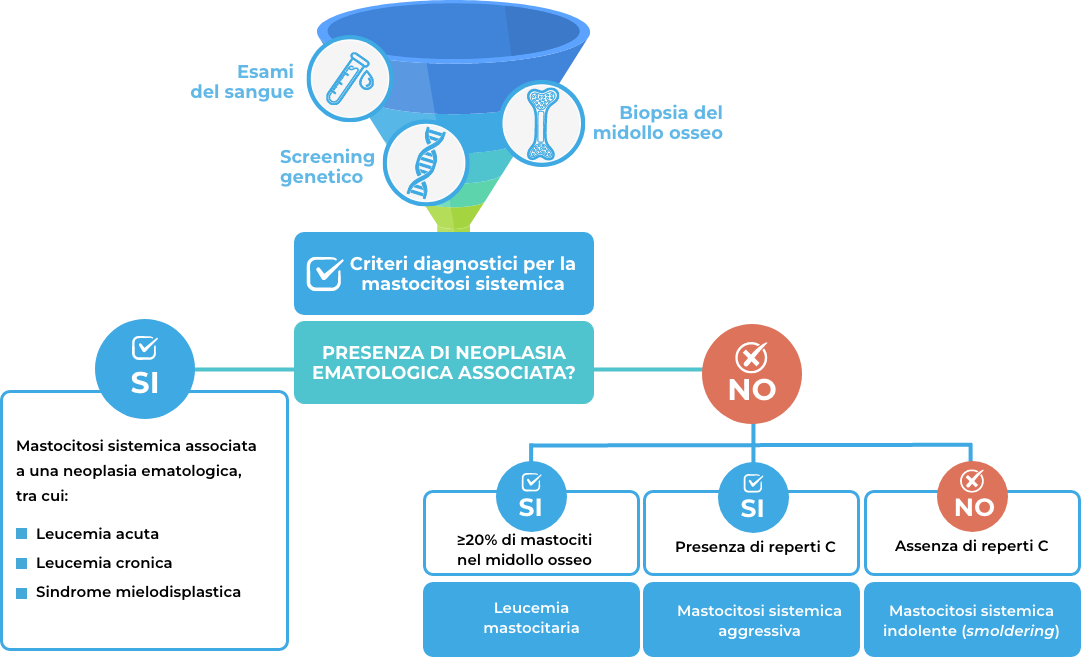
I risultati dei vari test sono utilizzati per valutare la corrispondenza con i criteri di identificazione della mastocitosi sistemica.
Quali sono i criteri diagnostici stabiliti dall’Organizzazione Mondiale della Sanità?
La diagnosi di mastocitosi sistemica è confermata quando sono presenti:
- il criterio maggiore e 1 criterio minore,
- almeno 3 criteri minori.

*Le biopsie sono prelevate da sezioni di midollo osseo e/o da organi extracutanei
+Questo parametro non è considerato valido in presenza di una patologia mieloide clonale associata
Vediamo insieme quali sono i test diagnostici che sono necessari per formulare una diagnosi di mastocitosi sistemica.
Si tratta di 3 analisi di laboratorio:

1.
Istologia e
immunofenotipizzazione
sul midollo

2.
Misurazione
dei livelli
di triptasi sierica

3.
Test di
genetica molecolare
Dopo aver diagnosticato la mastocitosi sistemica, l’esatta tipologia può essere definita attraverso criteri che valutano:
il carico di malattia, mediante reperti B
- biopsia del midollo osseo con infiltrazione ≥30% di mastociti,
- segni di displasia o mieloproliferazione, ingrossamento del fegato e/o ingrossamento palpabile della milza,
- organomegalia.
l’aggressività della malattia, mediante reperti C
- la presenza di ≥1 reperto C è indicativa di mastocitosi sistemica aggressiva. In questo caso è necessario iniziare una terapia di citoriduzione;
- l’assenza di reperti C è indicativa di mastocitosi sistemica indolente o mastocitosi sistemica smoldering.
L'identikit
La classificazione dell’Organizzazione Mondiale della Sanità identifica differenti tipologie di mastocitosi
sistemica avanzata:
mastocitosi sistemica con una neoplasia ematologica associata (SM-AHN)
- soddisfa criteri per la mastocitosi sistemica*,
- è presente un altro disturbo clonale ematico non derivato da mastociti,
- è solitamente osservata in concomitanza con ASM o neoplasie mieloidi.
mastocitosi sistemica aggressiva (ASM)
- mastocitosi sistemica* con ≥1 reperto C,
- nessuna evidenza di leucemia mastocitaria.
leucemia mastocitaria (MCL)
- diffusione leucemica di mastociti,
- ≥20% di mastociti nel midollo osseo e ≥10% di mastociti nel sangue periferico.
*la mastocitosi sistemica colpisce ≥1 organo extracutaneo, tra cui midollo osseo, tratto gastrointestinale, linfonodi e milza, con o senza coinvolgimento cutaneo.
Trattamento
Il trattamento della mastocitosi sistemica è personalizzato sul singolo Paziente in base a:
- gravità dei sintomi,
- manifestazioni della malattia,
- decorso clinico e prognosi.
Dal momento che non esiste un trattamento farmacologico in grado di far guarire dalla mastocitosi, le soluzioni terapeutiche a disposizione hanno un triplice obiettivo:
- ridurre i sintomi, andando a controllare la secrezione e gli effetti dei mediatori mastocitari,
- diminuire l’entità dell’infiltrazione dei mastociti con terapie di citoriduzione,
- trattare le complicanze, come la disfunzione d’organo, causate dall’infiltrazione dei mastociti.
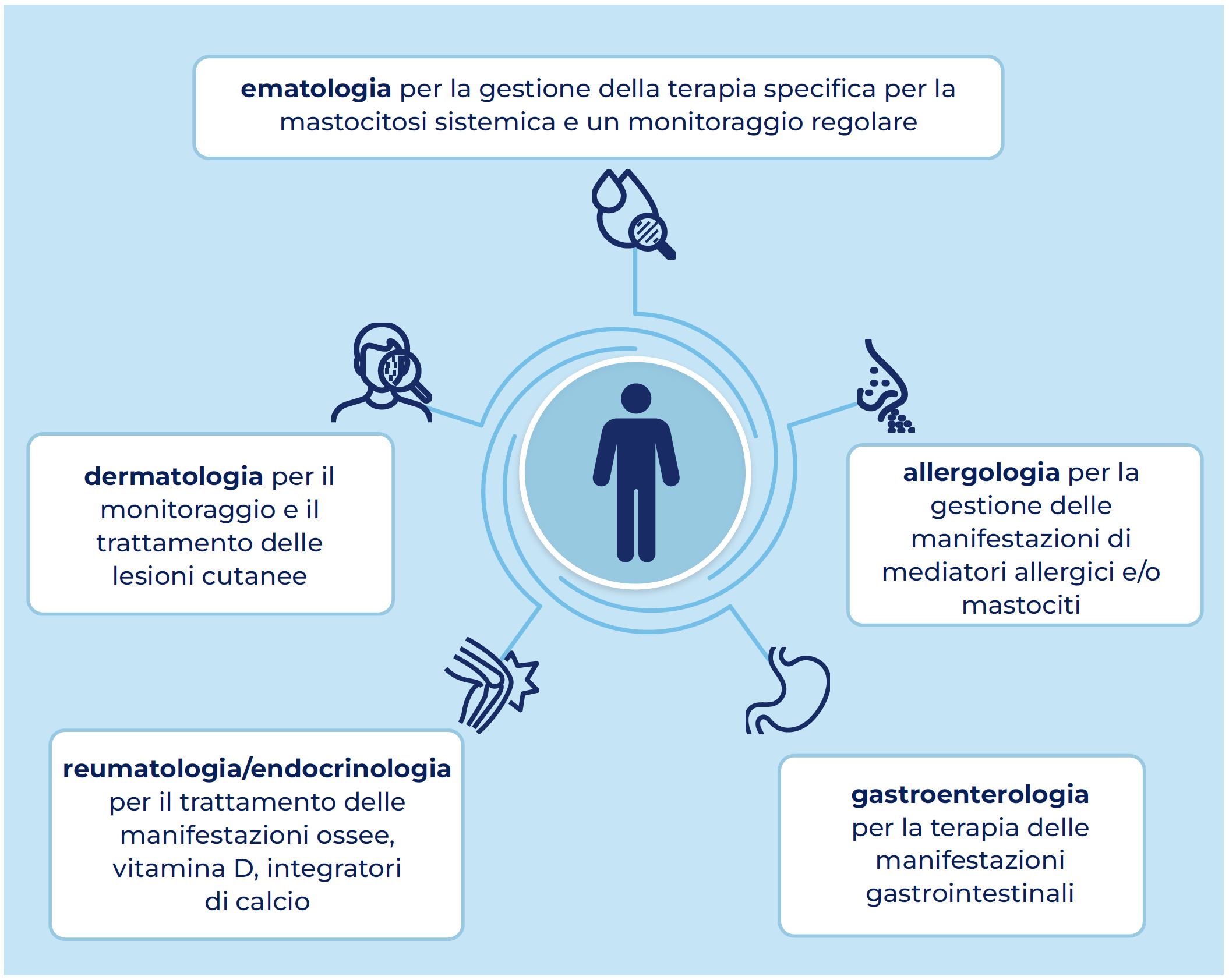
Data la complessità della malattia, il trattamento e la gestione del Paziente con mastocitosi necessitano del coinvolgimento di più specialità mediche:
Le opzioni di trattamento ad oggi disponibili possono essere classificate in:
- terapia anti-mediatore per ridurre il rilascio o gli effetti dei mediatori dei mastociti,
- terapie citoriduttive volte a ridurre il carico di mastociti.
Tra le opzioni terapeutiche a disposizione per la mastocitosi sistemica in fase avanzata si hanno:
- inibitori del recettore C-KIT,
- chemioterapici,
- terapia antinfiammatoria,
- terapie di supporto alla sintomatologia.
REFERENZE
- Allergy, Volume: 77, Issue: 1, Pages: 83-99, First published: 06 May 2021, DOI: 10.1111/all.14881.
- Arber DA et al. Blood 2016;127:2391-2405.
- Arock M et al. EurJ Haematol2015;94(6):474-490.
- Criscuolo M. et al. Oncol Ther 6, 129–140 (2018).
- Fuller SJ. Hematol Oncol Clin NAn. 2012;6:1143-1158.
- Garcia-Montero AC et al. Blood.2006;108:2366-2372.
- Gotlib J et al. Blood. 2010;116 [abstract 316].
- Horny H-P et al. Mastocytosis. In: SwerdlowS, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC; 2008:54-63.
- Horny HP et al. J Clin Pathol. 2004;57:604-8.
- Khoury JD et al. Leucemia. 2022 Luglio;36(7):1703-1719.
- Kristensen T et al. J MolDiagn. 2011;13:180-188.
- Lim KH et al. Blood. 2009; 113:5727-36.
- Longley BJ et al. Leuk Res. 2001;25:571-576.
- Orfao A et al. Br J Haematol.2007;138:12-30.
- Pardanani A. Am J Hematol. 2013.88:613-624.
- Pardanani A. Am J Hematol. 2015.90:251-262.
- Pieri L. Am J Hematol, 2016 Apr 7. Epub ahead of print.
- Roskoski R Jr. Biochem Biophys Res Commun. 2005;338:1307-1315.
- Sánchez-Muñoz L et al. Mod Pathol. 2011:24:1157-68.
- Sperr WR, Valent P. Expert Rev Hematol. 2012;5:261-274.
- The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms.
- Valent P et al. 2021 Ottobre 13;5(11):E646.
- Valent P et al. Int J Mol Sci. 2019 Jun 18; 20(12):2976.
- Wang SA et al. Am J Hematol: 2013;88:219-24.
- Wimazal F et al. Int Arch Allergy Immunol. 2012; 157:399-405.
- Zanotti R et al. Mediterr J Hematol Infect Dis. 2021 Nov 1;13(1):e2021068.